



Disease of the Month – Gaucher Disease
Disease of the Month – Gaucher Disease
Shots:
- Gaucher disease is a rare, autosomal recessive disorder that enlarges organs like the bone marrow, liver, and spleen by depositing fats (lipids) in them
- In this reprise of our Disease of the Month report, we present an enlightening description of Gaucher disease with a thorough study of epidemiology, market size, disease management, potential medicines, patient advocacy groups, and the major key players involved
- For a deep dive landscape, reach out to us at connect@pharmashots.com
Gaucher disease is a lysosomal storage disorder (LSD) in which the deficiency of the enzyme glucocerebrosidase leads to excessive accumulation of glycolipid glucocerebroside in the body, primarily in bone marrow, liver, and spleen. This condition leads to the enlargement of the liver and spleen. There are five subdivisions of the disease:1,2
- Type 1 (Non-Neuropathic): It is characterized by the clinical or radiographic signs of bone disease hepatosplenomegaly, anemia, thrombocytopenia & lung disease without CNS illness
- Type 2 (Acute Neuropathic Form): A rapidly progressive form that causes main CNS illness, beginning before the age of 2 years. It minimizes psychomotor development & leads to death around the ages of 2 & 4 years
- Type 3 (Chronic Neuropathic Form): A slow progressive form that causes major CNS disease, beginning during childhood. People may survive into their 20s & 30s
- Perinatal-lethal form: It is associated with ichthyosiform or collodion skin abnormalities, or nonimmune hydrops fetalis
- Cardiovascular form: It is defined by calcification of the aortic and mitral valves, mild splenomegaly, ocular opacities, and supranuclear ophthalmoplegia
Symptoms 3,4
Symptoms of Gaucher Disease may vary from person to person. Some of the commonly seen symptoms are:
- Enlarged spleen and liver
- Eye mobility disorders
- Yellow dots on the eyes
- Anemia
- Fatigue
- Bruising and weak bones
- Lung issues
- Seizures
Causes 1,5,6
- Genetic Mutation: Mutations in the GBA1 gene, which encodes the glucocerebrosidase enzyme, lead to reduced activity or production of the enzyme
- Autosomal Recessive Inheritance: A person must inherit two copies of the mutated gene to develop the disease. Individuals with only one mutated gene are carriers and usually are asymptomatic
- Enzyme Deficiency: Deficiency of glucocerebrosidase leads to the buildup of glucocerebroside in the lysosomes of cells, causing organ enlargement
Diagnosis 5
- Enzyme Testing: A blood test known as beta-glucosidase leukocyte (BGL) test to examine enzyme activity
- Genetic Testing: A blood or saliva sample is used for genetic testing to identify genetic mutations
- Newborn Testing: mainly done by blood testing
- Bone Marrow Testing: Bone marrow testing is carried out to rule out the possibilities of leukemia and hematological cancers
Management and Treatment 5
Type 1 Gaucher disease is treatable with enzyme replacement therapy or substrate reduction therapy that either boosts enzyme levels or reduces the fatty substance accumulation in the body. However, there is no treatment for the neurological damage associated with Gaucher disease types 2 and 3
Treatment options for Gaucher disease type 1 include:
- Enzyme replacement therapy (ERT): Administered as an IV infusion, ERT restores low GCase enzyme levels with a modified version, enabling the breakdown of glucocerebroside, a fatty substance that accumulates in organs and bone marrow
- Substrate Reduction Therapy (SRT): Administered as an oral medication, SRT lowers the body's glucocerebroside rather than restoring GCase enzyme levels. This reduction minimizes the workload on the body’s enzymes by providing less glucocerebroside for breakdown
- Symptoms Treatment: Alongside ERT and SRT for enzyme deficiency and glucocerebroside buildup, additional treatments may be necessary for Gaucher disease symptoms and complications. These include:
- blood transfusions for severe anemia
- medications for bone pain
- orthopedic surgeries like joint replacement
Epidemiology: 7
The frequency is around 1/100,000, with an annual incidence among the general population of roughly 1/60,000. However, 1/1,000 Ashkenazi Jews can be affected by GD
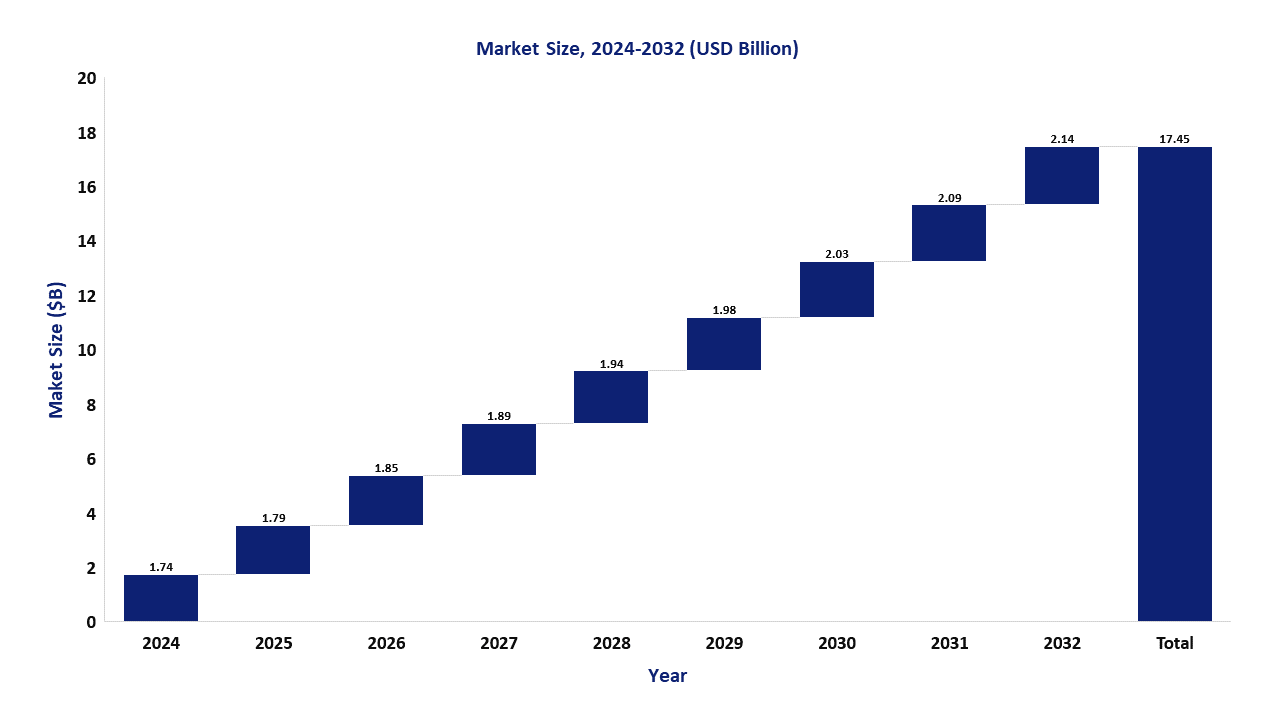
Market Size: 8
The global Gaucher Disease market size was valued at $1.69B in 2023 and is forecasted to reach a value of $2.14B by 2032 at a CAGR of 2.6% between 2023 and 2032
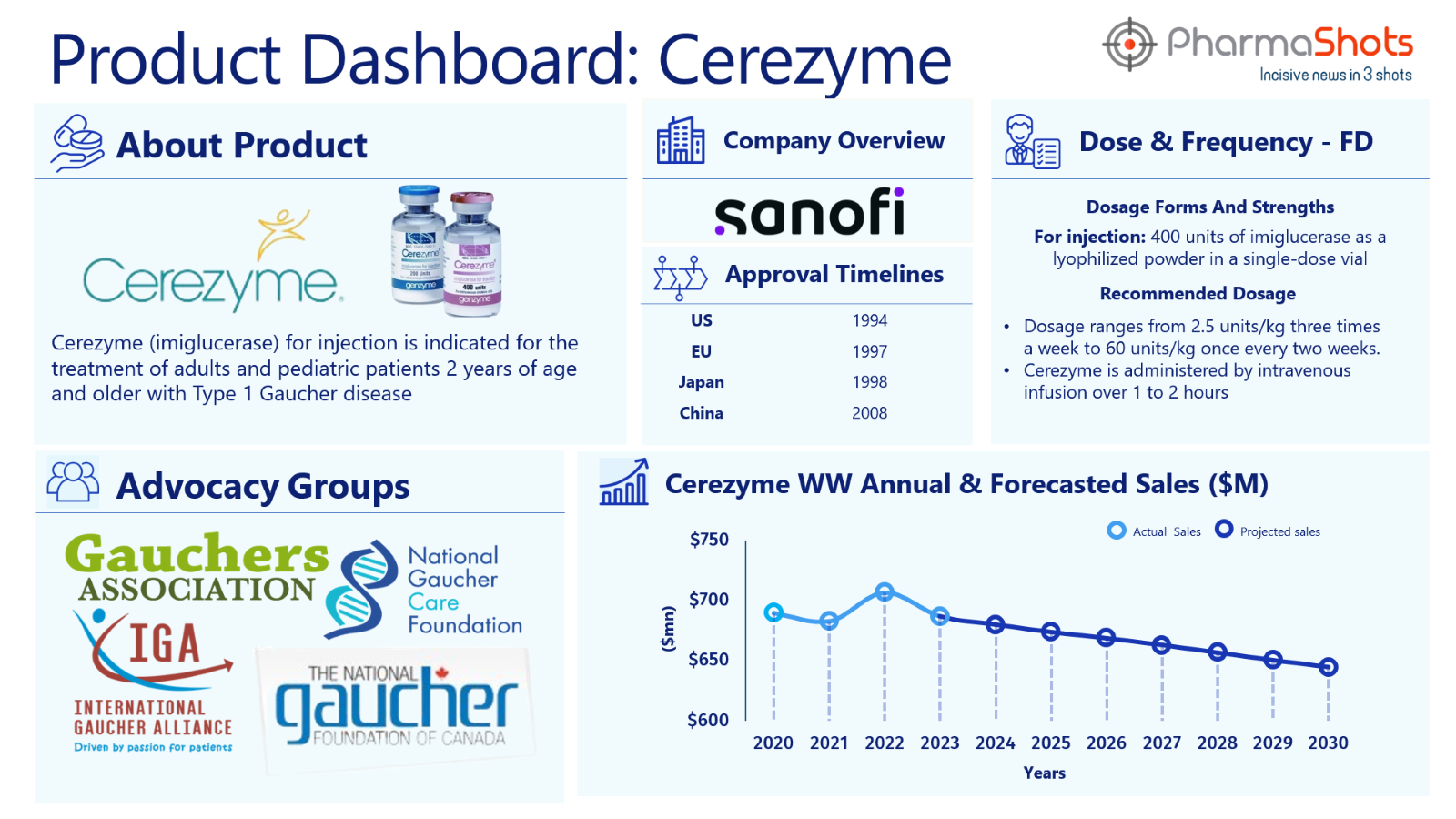
Product Dashboard:
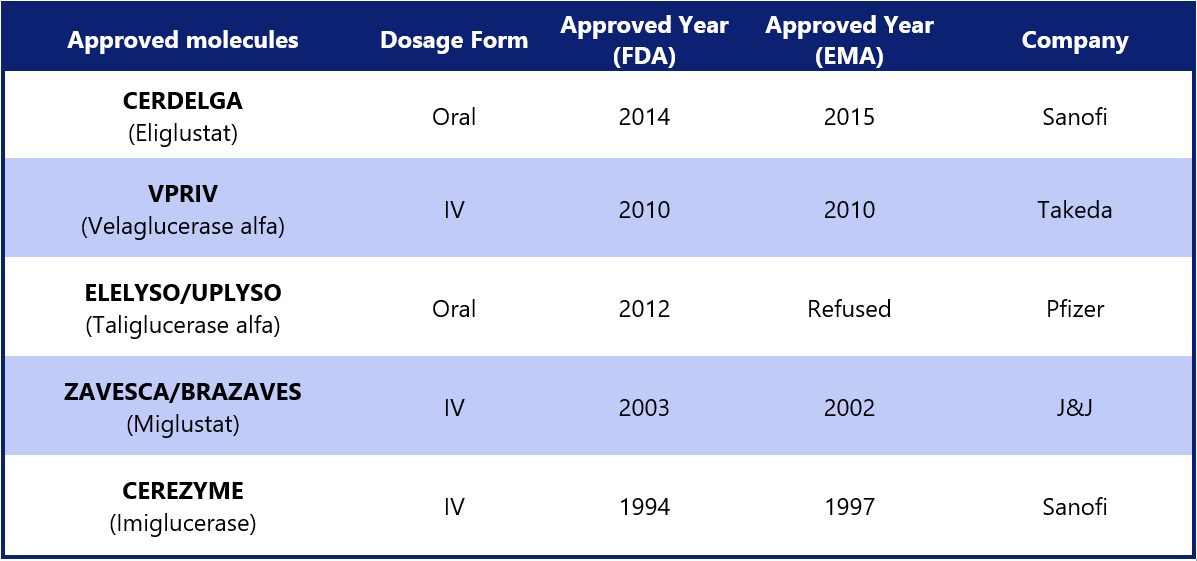
Key Players in the Market:
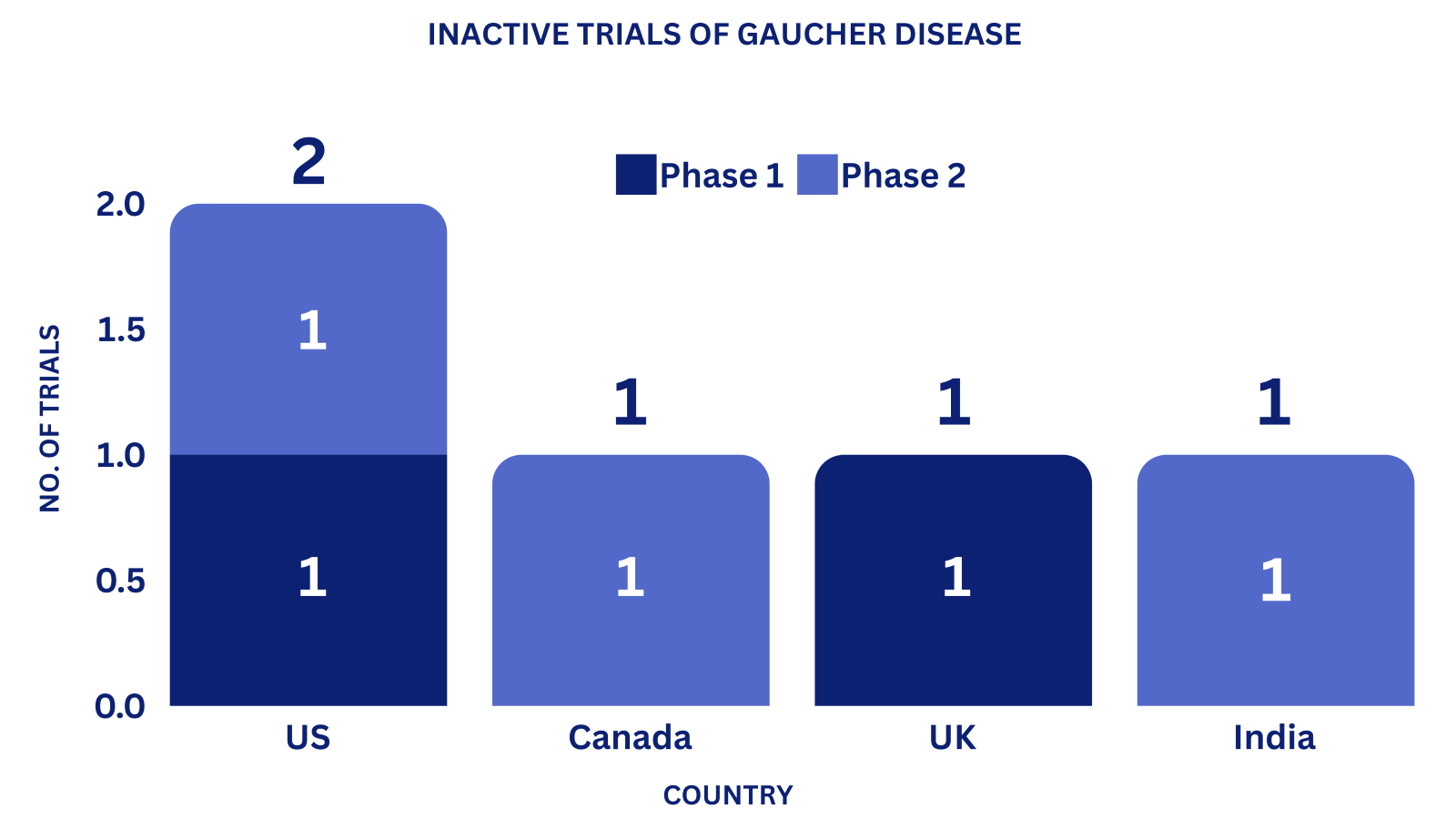
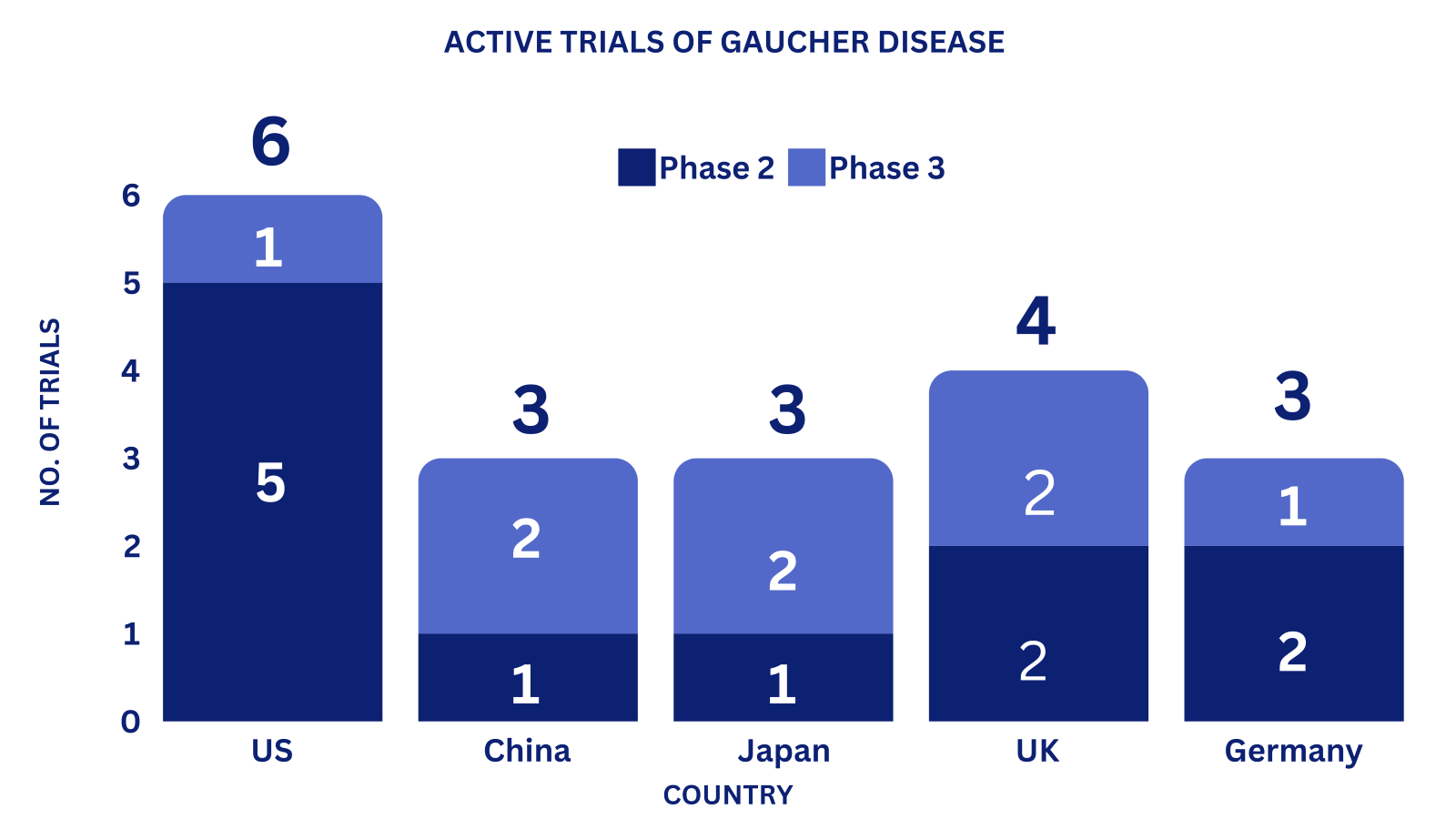
Clinical Trial Analysis: 9
Based on the geographical distribution, the interventional and industry-sponsored clinical trials are classified in the below-mentioned graph into two groups based on their status: Active (recruiting; active, not recruiting; not yet recruiting; enrolling by invitation and suspended) and Inactive (withdrawn; terminated and trials with unknown status)
The maximum number of active trials is being conducted in the US, UK, China, Japan, and Germany (as represented in the graph)
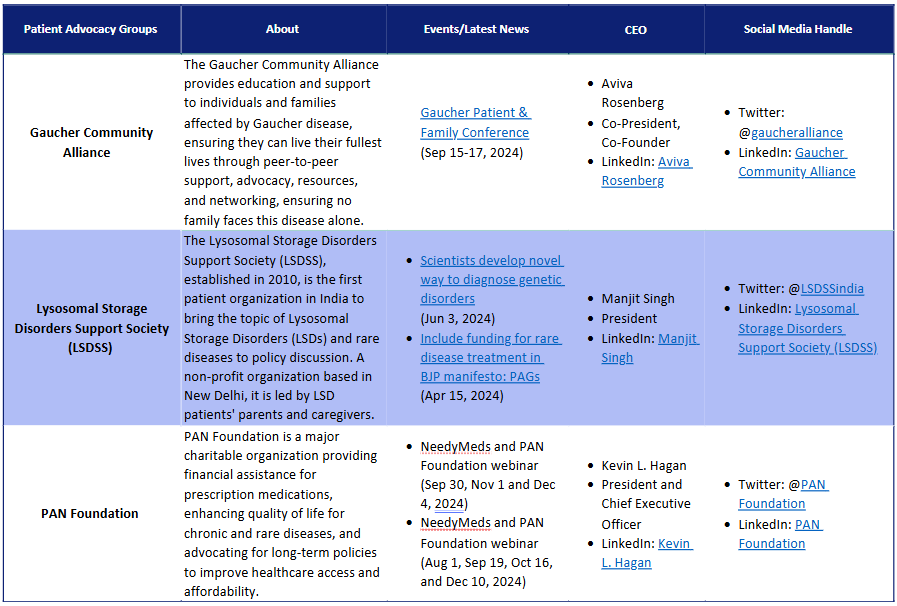
Patient Advocacy Groups (PAGs) for Gaucher disease:
Patient advocacy for Gaucher Disease can be challenging owing to low prevalence, and limited patient care. Though PAGs are dedicatedly working to address the problems of Gaucher Disease patients and their families.
Patient Stories 10,11
Lauren 10
At the age of 18, I never thought routine blood drawn for acne medication would lead to a life-changing diagnosis. When my iron levels came back low, I was sent to a hematologist who prescribed iron pills. A year later, my blood counts hadn't improved, and my doctor suggested a bone marrow biopsy. The results revealed Type 1 Gaucher disease, a rare genetic disorder.
Living with GD for the past five years has been challenging, but I've learned to manage my symptoms and live a fulfilling life. It's been a journey of self-discovery and resilience, navigating a world of doctor's appointments and treatment plans. Despite the initial shock of the diagnosis, I've found strength in knowing what I'm up against and taking control of my health.
It all started with a simple blood draw, but it led me down a path of understanding and acceptance.
Brian 11
When I was four, I was diagnosed with Type 1 Gaucher disease. My condition was severe, and the prognosis looked bleak. But my parents refused to accept this fate. They sought out Dr. Roscoe Brady, a pioneering researcher in Gaucher disease treatment. Fortunately, I was deemed an ideal candidate for clinical trials, and Dr. Brady enrolled me in an experimental program that would change everything.
I became the first person in the world to successfully receive enzyme replacement therapy for Gaucher disease. That groundbreaking treatment not only transformed my life but also opened doors for countless others facing the same battle. Today, I lead the National Gaucher Foundation, using my experience to advocate for and support others affected by this disease. My journey has become a testament to hope and resilience, and I'm passionate about helping others find their path to healing.
References
- NIH StatPearls
- NIH GeneReviews
- Hopkins medicine
- Mayo Clinic
- Gaucherdisease.org
- Rarediseases.org
- Orpha.net
- Grand view research
- ClinicalTrials.gov
- Patient Story 1
- Patient Story 2
Related Post: Disease of the Month – Small Cell Lung Cancer
Tags

An avid reader and a dedicated learner, Prince works as a Content Writer at PharmaShots. Prince possesses an exceptional quality of breaking down the barriers of words by simplifying the terms in digestible chunks to make content readable and comprehensible. Prince likes to read books and loves to spend his free time learning and upskilling himself.










